Ion Channels: Structure and Function
1. Introduction
Ion channels are membrane protein complexes and their function is to
facilitate the diffusion of ions across biological membranes. Membranes,
or phospholipid bilayers, build a hydrophobic, low dielectric barrier
to hydrophilic and charged molecules. They are electrical insulators.
Ion channels provide a high conducting, hydrophilic pathway across the
hydrophobic interior of the membrane. The channel, or pore structure,
is said to catalyze the 'reaction' of transporting charged molecules across
a low dielectric medium. The 'catalytic site', the central channel, is
either open or closed. The open channel conformation can
be compared to the transition state of the enzyme-substrate complex, where
ions are tightly associated to the catalytic site. The conformational
change between closed and open state is called gating, as in opening
and closing a gate. Channel gating is controlled by external factors like
enzymes are controlled by modulators and effectors. Ion channels can be
classified according to which chemical or physical modulator controls
their gating activity. Thus we have different groups of channels as
summarized below:
- ligand gated channels neurotransmitters- voltage gated channels transmembrane potential (electric field)
- second messenger gated channels nucleotides, G-proteins
- mechanosensitive channels osmotic pressure, membrane curvature
- gap junctions, porins not gated
Channels are also ion selective. They can discriminate between
size and charge of the permeant molecule. All ion channels are complexes
of transmembrane proteins, sometimes they contain cytoplasmic subunits,
often they are glycosylated. The 3-D structure of most ion channels, is
not known, with two notable exceptions, porins and a K-channel, both of
bacterial origin. There exists, however, a multitude of biochemical and
functional data, combined with mutagenesis experiments that give information
about the transmembrane topology of these proteins, dividing it
into transmembrane segments and extramembraneous loops/domains. Often
size and location of loops on one side or the other of the membrane can
be determined by chemically modifying the protein and analyzing which
amino acids have been modified.
2. Bacterial Porins
The first structure (in 1990) of an ion channel solved at atomic resolution <0.3nm is that of the bacterial porins, a family of homo-trimeric channel proteins. Each subunit contains 16 to 18 transmembrane, anti-parallel b -strands forming a b-barrel structure. The b-strands are amphipathic, they contain alternating polar and non-polar residues and the inter-strand interaction is fully saturated with H-bonding. This creates a hydrophilic pore interior providing a water filled channel. The channel has a large diameter of 0.8x1.1nm, and is non-selective for small ions. However, it has an upper exclusion size limit corresponding to molecular weights of about 600 Dalton of permeants. Because most metabolites have molecular weights lower than 600 Da and have been shown to pass through porin channels, porins are called general diffusion pores.
Fig. Ribbon diagram of porin barrel and projection of outer face of this barrel
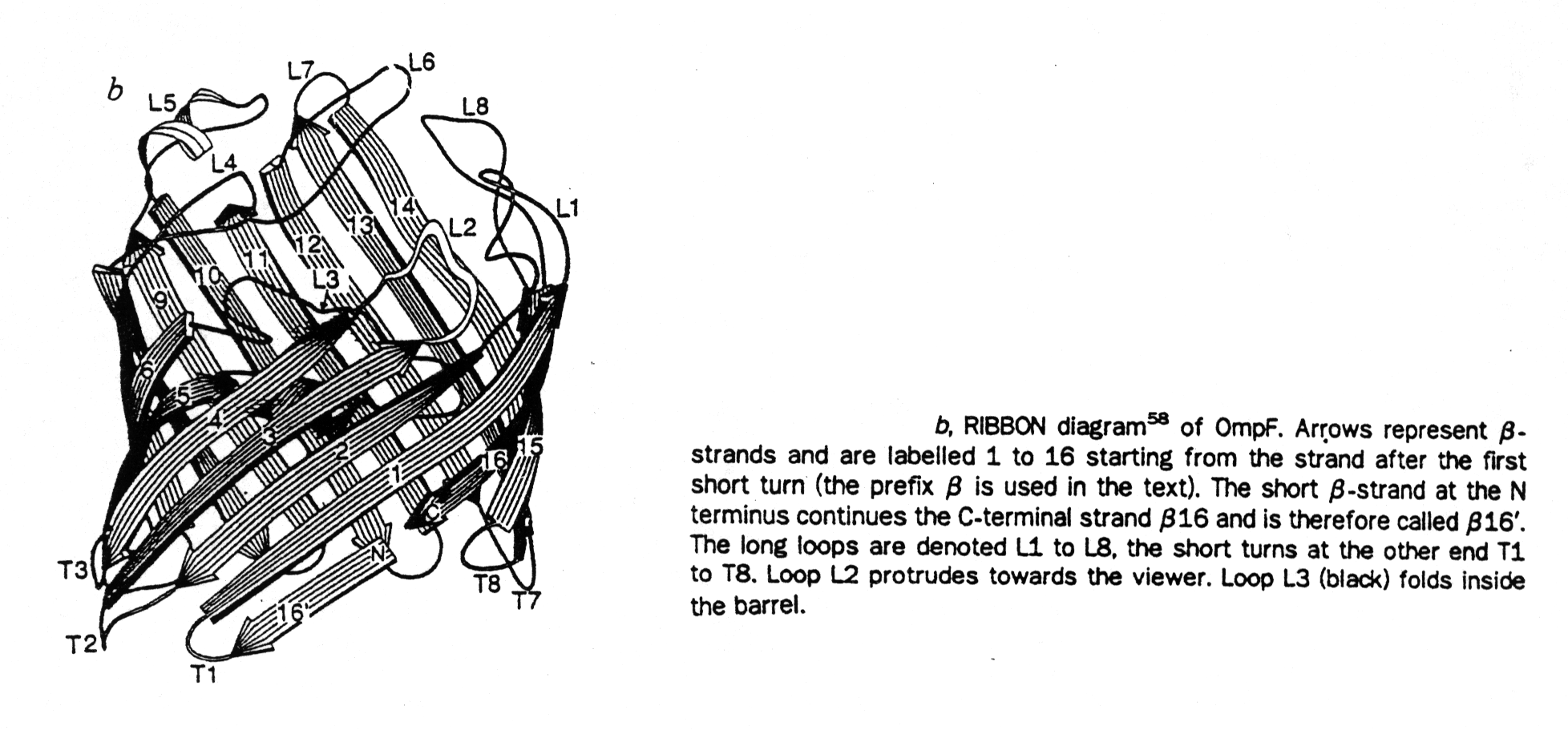
from Cowan et al., 1992
Fig. Projection map of porin barrel
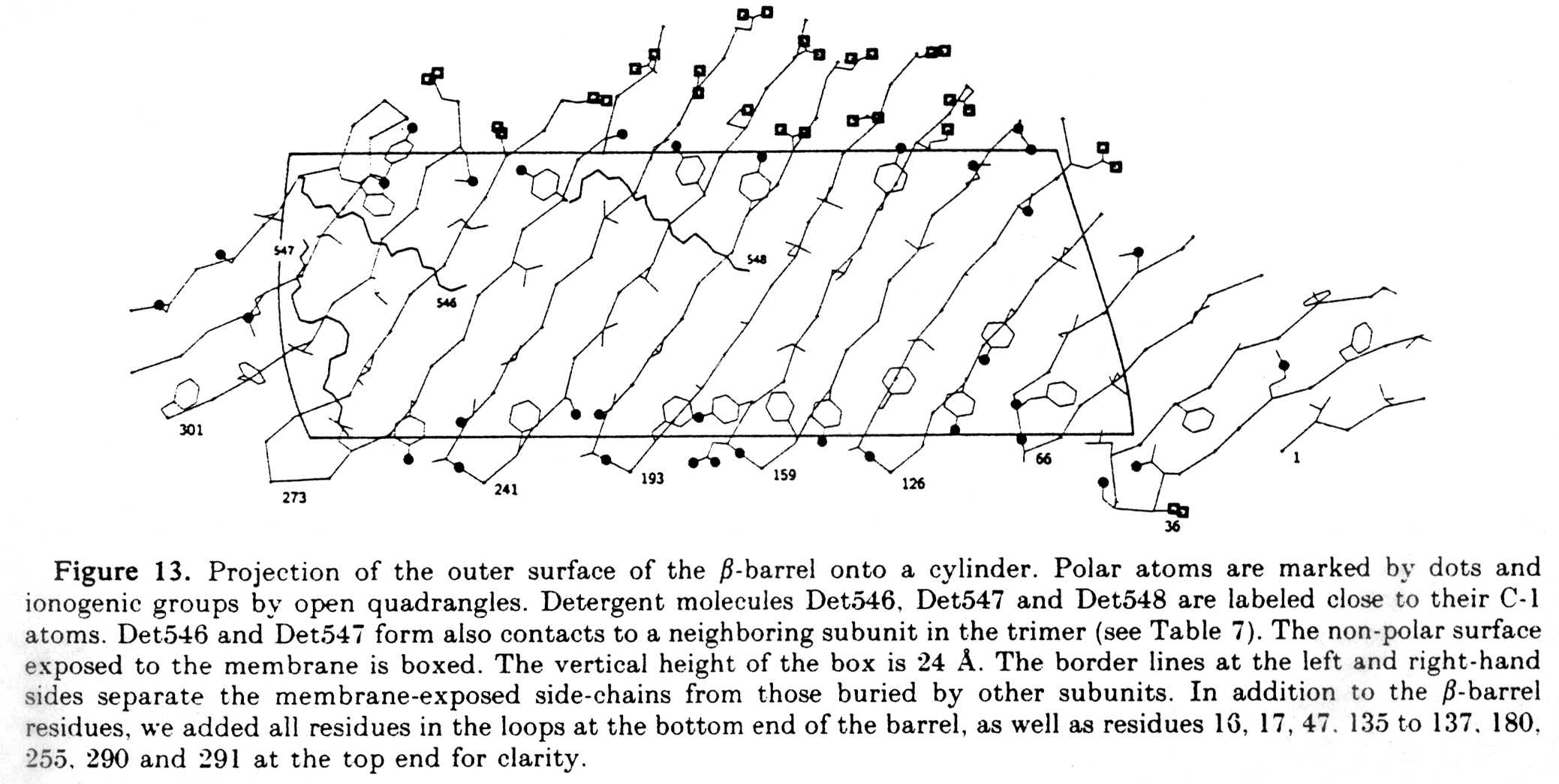
from Weiss and Schulz, 1992
3. Nicotinic Acetylcholine Receptor - nAChR
The structure of another ion channel, the nicotinic acetylcholine receptor, has been determined to 0.9nm resolution by cryo-electron microscopy. The nAChR is a heteromeric glycoprotein complex composed of five integral membrane proteins in a stoichiometry of a2bgd.
Table Subunits of the nicotinic acetylcholine receptor
|
|
metry |
acids |
weight |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
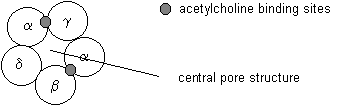
The five subunits are arranged in a circular fashion around a central hole that provides an ion pathway across the post-synaptic cell membrane. The pentameric complex has a five fold pseudo-symmetry because its subunits are not identical. The receptor complex binds two molecules of acetylcholine. Acetylcholine binding induces the opening of the channel. Coupling between the two ACh binding sites and the channel gate is an allosteric mechanism because the binding of a ligand causes a structural change on a distant place in the receptor unit.
Fig. Pentameric arrangement of nAChR subunits
The transmembrane topology of the AChR has been inferred from hydrophobicity analysis and secondary structure prediction of the primary sequence. So called hydropathy plots indicate the grouping of hydrophobic stretches in the sequence that are identified as spanning the membrane, usually in the form of a-helices. Four transmembrane segments have been proposed for the nAChR subunits, and the orientation of the receptor within the post-synaptic membrane has been modeled has having a large N-terminal domain outside the cell facing the synaptic cleft. This is also the location of the agonist binding site in the alpha subunits discussed above.
Fig. Two hydrophobicity scales of amino acids
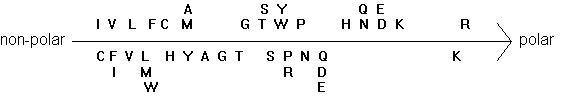
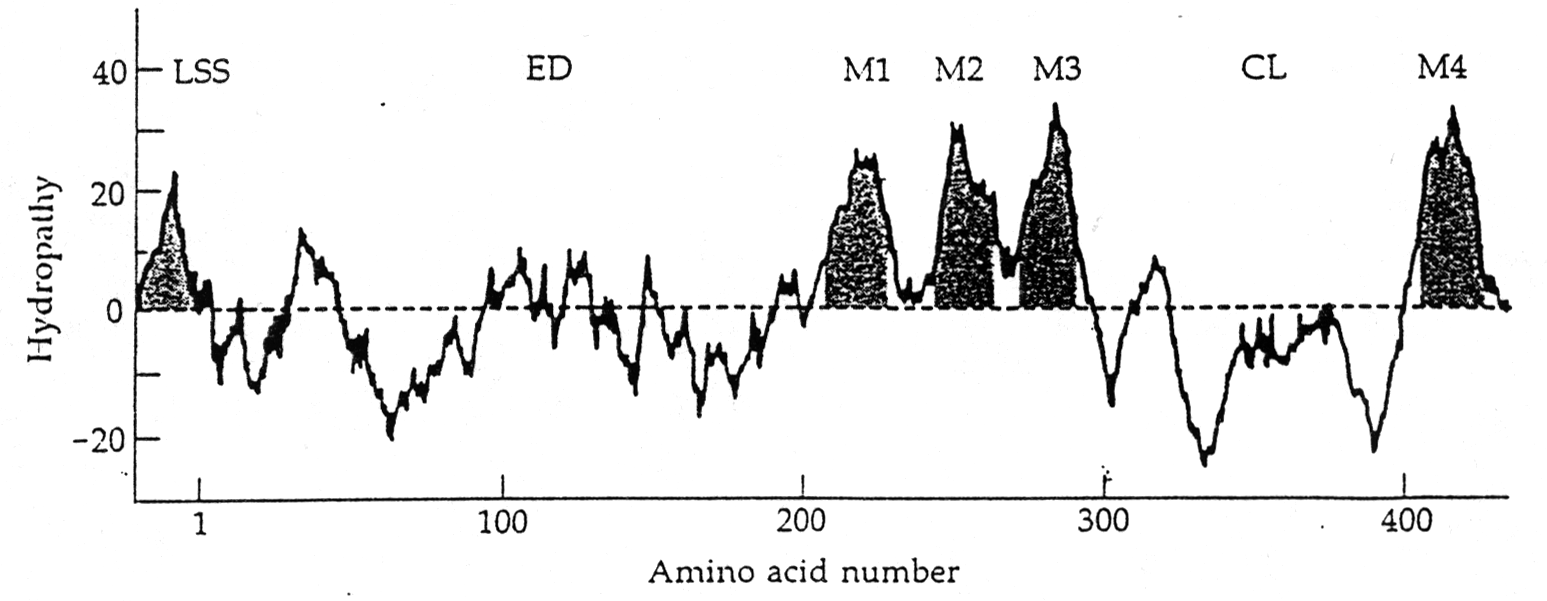
Electron microscopy providing a high resolution of 0.9nm shows the presence of such an a-helix surrounding the central pore. It also shows that the helix is found in a linear form when the channel is open, but exhibits a kink in the middle of the membrane when the channel is closed. A leucine residue in each subunit is located at this kink. The large part of the membrane buried structure, however, does not seem to exhibit any a -helices. It has tentatively been proposed (but not shown) to form a b-barrel structure giving the nAChR pentamer a similar membrane anchoring structure as found in porin trimers.
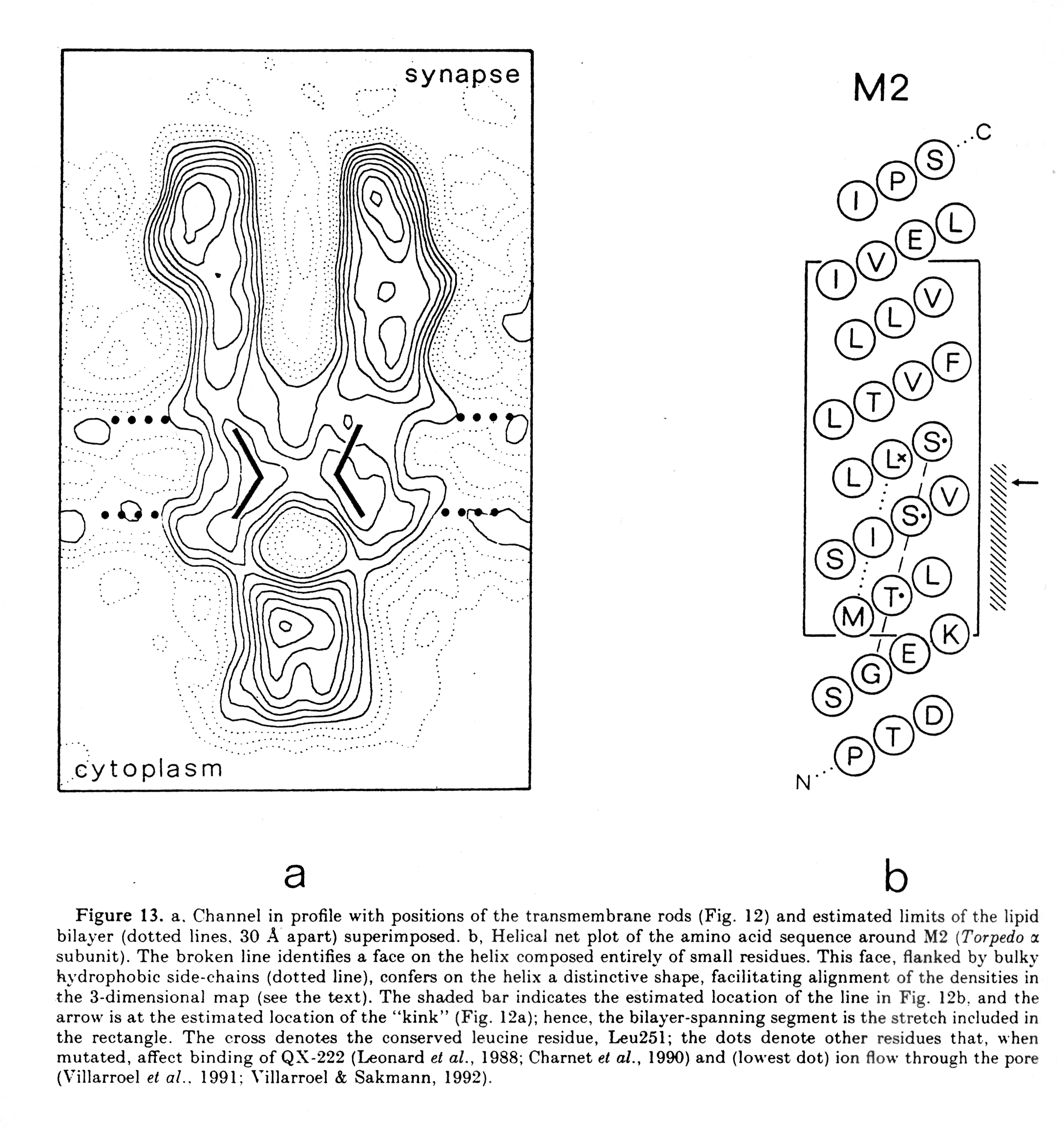
(from Unwin, 1993)
4. The acetylcholine binding site
Acetylcholine is a neurotransmitter and the natural agonist of the acetylcholine receptor.
Fig. Chemical structure of acetylcholine
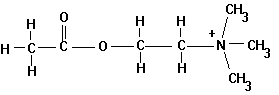
Besides ACh there are many other agonists that are important pharmacological agents or drugs that affect the activity of the receptor. The most important one is nicotine, which also gave this receptor type its specific name, nicotinic acetylcholine receptor. Agonists are positive modulators of the receptor activity. Antagonists are molecules that have the opposite effect of agonists, they inhibit receptor activity. The most commonly known inhibitors are the curare alkaloids and a-bungarotoxin, a small protein that irreversibly binds to the agonist binding pocket thus inactivating the receptor by blocking access for the agonist.
There are two ACh binding pockets in the receptor complex located in the clefts (subunit interface) between the a subunits and the g and b subunits respectively. Two molecules of ACh must bind to activate the receptor, i.e., to open the channel for ions to flow across the membrane. Labeling experiments, where small detector molecules can be covalently linked to amino acid side chains, showed that 5 residues in the N-terminal part (extracellular) of the a subunits are involved in agonist binding. These are residues Tyr93, Trp149, Tyr190, Cys192, and Cys193. The location of the amino acids in the polypeptide indicates that loops from different sites in the sequence are forming the binding pocket, similar to the catalytic triade of serine proteases and the heme binding pocket in globins.
The mechanism to open the channel is a long range effect of a conformational change that starts out as a small local conformational change as described for the oxygen binding to the heme group in globins. The distance between two heme binding sites in hemoglobin is about 2.5nm and the distance between the acetylcholine binding sites and the channel gate (distance between ACh binding site and membrane), the structure in the complex that opens and closes the ion pathway across the membrane, is about 2.5nm as well. This allosteric mechanism can be summarized in the following reaction scheme:
CLOSED R + A <=> RA + A <=> RA2 <=> R*A2 OPEN
The scheme shows that the combined effect of two bound agonists brings the receptor into its transition, or open state (R*), where the receptor exhibits an equilibrium between the closed and open channel. This equilibrium is measured as open probability of the channel P(open).
In addition to the kinetic scheme of ligand binding and channel opening,
the receptor can switch into a desensitized state, i.e., a conformation
where the channel is closed in the presence of two bound acetylcholine
molecules. The channel is unable to reopen until the ligands first dissociate
from their binding site. It could be shown that the affinity of the ligand
to the receptor in the desensitized state, D, is much higher than in the
normal, open or closed state R therbey preventing the postsynaptic membrane
from overstimulation.
5. K-channel structure
A. Non-voltage-gated
The crystal structure of a non-voltage gated, two-transmembrane spanning K-channel from the bacteria Streptomyces lividans (KcsA K+ channel) has been solved to 3.2 angstrom resolution (amino acids 126-158 at the carboxy terminal end have been cleaved off). Like several other membrane proteins (see projection map for porin barrel), it has two rings of aromatic amino acids positioned to extend into the lipid bilayer, presumably near the membrane-water interfaces. A subunit is inserted into the tetramer such that one transmembrane helix (inner helix) partially facing the central pore (45Å long) while the other (outer helix) faces the lipid membrane. The inner helices are tilted with respect to the membrane normal by about 25° and are slightly kinked with the wider part facing the outside of the cell allowing the structure to form the pore region near the extracellular surface of the membrane. This region contains the K+ channel signature sequence, which forms the selectivity filter. Within the selectivity filter the side chain orientation preclude their participation in ion coordination, leaving this function to the oxygen atoms of the main chain carbonyls. They are forming an oxygen ring coordinating a dehydrated K+ ion. The K+ ion thus has only a very small distance to diffuse from one site to the next within the selectivity filter.
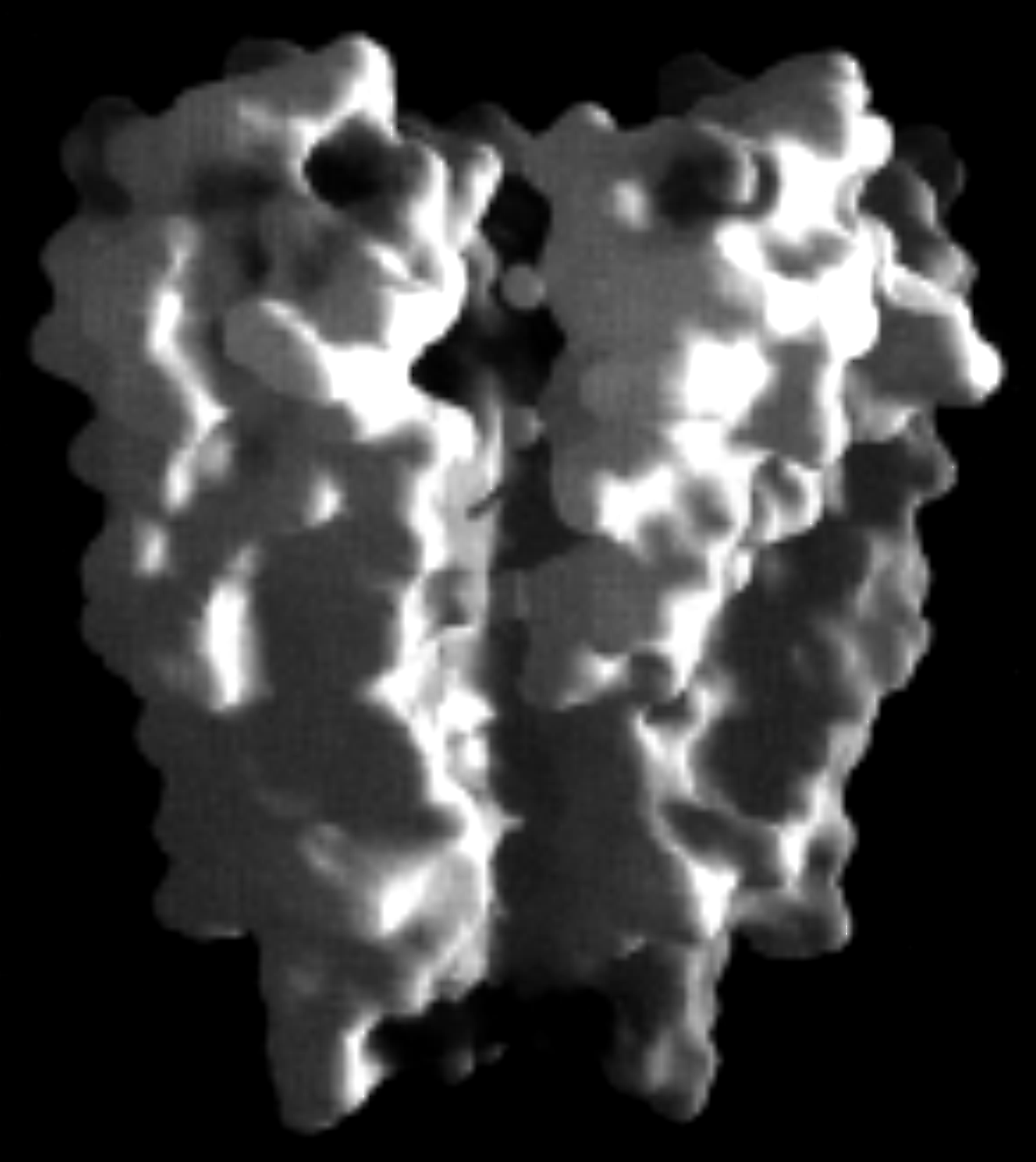


electrostatic surface
pore volume
selectivity filter & ion binding sites
(from Doyle, 1998)
The crystal structure includes permeating ions (Rb+ or Cs+) and shows three binding sites. One of three cations is stabilized in the middle of the membrane within an aqueous cavity (10 Å diameter) at the negatively charged carboxyl end (helix dipole) of four central a -helices, one from each subunit. Because of the size of the cavity, this central ion is proposed to be hydrated. Furthermore, the structure shows that, with the exception of the selectivity filter (12Å long), the pore lining is mainly hydrophobic (cytoplasmic side of channel lumen, corresponds to lower half of pore in above figure), a general property of K-channels. This might explain why most K-channels have a very high flux rate, or ion conductance.
B. Voltage-gated
Potassium channels form a family of K+ selective, voltage-gated channels in excitable membranes such as neuronal and muscle cell membranes. So far over 50 different genes have been cloned and sequenced from mammals, plants, and microorganisms. Most potassium channels form homo- or hetero-tetrameric protein complexes with each subunit having 6 transmembrane segments (S1 - S6) and a pore loop structure connecting the fifth and sixth segments. Transmembrane segment S4 contains a series of positively charges amino acids which have been shown to be essential for voltage-sensing. Although the crystal structure of KcsA is a non-voltage-gated type, the sequence similarities of the pore loop strongly suggest that all potassium channels have the same quaternary pore architecture.
6. Measuring single channel activity
How are ion channels studied experimentally? Ion channels catalyze the diffusion of ions across membranes with electrical currents in the order of pico Amperes (10-12A). The recording of single channel currents shows two current levels corresponding to the closed and open state respectively. Transitions between these two states are very fast and in the order of fractions of a millisecond, and appear in the recordings as rectangular jumps from one level to the other. The amplitude of the jump corresponds to the current. The current can be normalized and expressed as resistance (or its inverse, the conductance) because it is proportional to the membrane potential as defined by Ohm's law (E = R*I). Channels have ion flux rates of up to 106 ions/second.
Normally, the channels stay open for only a fraction of seconds, allowing the flux of tens of thousands of ions through the pore. The activity of the channel can be quantified as open probability, the fraction of time it stays in the open conformation. The open probability of a channel can be compared to the fractional saturation of hemoglobin with molecular oxygen. Whereas in hemoglobin the fractional saturation is related to the saturation pressure of the oxygen (the ligand of the heme), the open probability is related to the concentration of the acetylcholine in the synaptic cleft.
Fig. Single channel activity and voltage dependence of a Na-channel (from Catterall, 1988)
