Protein Motion - Molecular Dynamics
1. Thermal motion
Proteins are not static and rigid structures. The polypeptide backbone and specially the side chains are constantly moving due to thermal motion or kinetic energy of the atoms (Brownian motion). Thermal motion - displacement of functional groups - is necessary for protein catalysis (high substrate specificity & transition state stabilization) and allostery (transmit information to other units in a protein complex).
When studying the structure of a protein, different techniques give different information about motion in macromolecules. X-ray crystallography allows a time averaged 'snapshot' of the structure, which appears as if it were frozen. Electron microscopy too gives fixed structures due to the long exposure time. NMR enables a time resolved, dynamic structure. Specially small domains and loops may fluctuate rapidly and are not resolved by crystallography, but may be accessible for NMR. Indirect evidence for flexible parts in 'frozen' structures comes from incomplete or unresolved parts of the protein. However, it does not give any information about the nature of this flexibility. From the point of view of a dynamic system, the native structure of a protein is a collection of conformational substates that have statistically equal stabilities.
Experimental evidence for the importance of molecular motion in catalysis
has been obtained using a small, globular protein, Ribonuclease A.
At temperatures below 212° K where the
fold is basically frozen but solute diffusion is still fast, substrate
does not bind nor does bound substrate dissociate (even within days).
At the slightly elevated temperature of 228°
K, however, where protein motion can be measured, one finds rapid binding
and unbinding of the substrate from the enzyme as determined by product
formation. What are the time scales and distances of these movements at
atomic level?
| Time scale | Amplitude | Description |
| short femto, pico 10-15 - 10-12s |
0.001 - 0.1 Å | - bond stretching, angle bending - constraint dihedral motion |
| medium pico, nano 10-12 - 10-9s |
0.1 - 10 Å | - unhindered surface side chain motion - loop motion, collective motion |
| long nano, micro 10-9 - 10-6s |
1 - 100 Å | - folding in small peptides - helix coil transition |
| very long micro, second 10-6 - 10-1s |
10 - 100 Å | - protein folding |
2. Molecular dynamics
One way to understand the motion in proteins is to use computer simulations called molecular dynamics. With this technique changes in protein conformation are calculated. Atoms constantly are in motion because of thermal agitation or vibration. The structure of the macromolecule keeps the atoms in place and restricts their motion. One can define degrees of freedom that indicate how flexible a local molecular structure is. Taking the molecule butane as an example, we find 3 bonds r1, r2, r3, 2 bond angles Q1, Q2, and a single dihedral (or torsion) angle F resulting in 6 degrees of freedom of this 4 carbon backbone structure.
Fig. Structure and structural parameters in butane
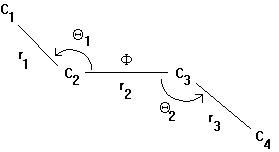
The forces to move atoms are divided into two categories:
hard degrees - small fluctuations: includes bonds (large force constant) and angles (smaller force constant)soft degrees - large fluctuation: includes torsion angles (smallest force constant)
In proteins we can state that, in general, bonds and angles can stretch
and bend slightly to greatly reduce the steric repulsion between atoms
C1 and C4 (in butane) during F
torsional transition. This describes the flexibility of the polypeptide
backbone which exhibits hard degrees of freedom and the two dihedral
angles F and Y,
as well as the side chain torsion angles cwhich
exhibit soft degrees of freedom in the unfolded protein. Folding
the polypeptide chain introduces hard degrees of freedom due to steric
hindrance between atoms (Van der Waals envelopes) in the side chains that
hinder the free rotation around the torsion angles. The large number of
H-bonds also restricts motion and introduces hard degrees of freedom (although
they have a smaller force constant than covalent bonds).
Molecular dynamics calculations use the above developed concept of degrees
of freedom of interaction between atoms in macromolecules. Thermal motion
produces a random walk of every atom in a structure and steric hindrance
restricts the calculation to allowed conformations as determined by the
torsion angle plot of proteins (Ramachandran plot). Molecular dynamics
is a process that calculates 'jumps' in protein conformational changes
on a very short time scale measure in nano- pico- or femto-second (10-12s).
The molecular dynamics calculations show motion in structures as a series
of snapshots of new, fixed conformations every ps. All calculations are
normally done in vacuum (dielectric constant 1-2) because the energy terms
of solvation and the number of solvent molecules make the calculation
yet too difficult.
3. Gramicidin A channel
To demonstrate protein motion we will look at a molecular dynamics calculation of the movement of a Na+ ion through a Gramicidin channel in vacuum. Gramicidin is a small 15 amino acid containing peptide that acts as an antibiotic or bactericide. It contains alternating L- and D-amino acids and adopts a right-handed helical conformation with backbone H-bonding similar to parallel b -strand interaction. The helix is therefore referred to as b-helix. The N- and C-terminal ends are covalently modified to remove the ionized groups to ease membrane incorporation and stabilize the functional state of the dimer channel. The cell-lytic activity of Gramicidin functions by incorporation into plasma membranes and dimer formation in a head-to head arrangement where the two polar, non-charged N-terminal ends sticking together. The 26Å long dimer helix spans the hydrophobic part of the membrane and provides an ion pathway for monovalent cations such as Na+, K+, Li+, Cs+, and Rb+. It excludes negative ions as well as divalent cations (Ca++).
The Gramicidin peptide is hydrophobic and all amino acid side chains stick away from the peptide backbone. The interior of the helix forms the channel with a Van der Waals diameter of 4Å. Ions passing through the channel have to strip of their hydration shell. As the hydration shell of Cl- is thermodynamically stronger than that of Na+, the channel can select for Na+.
Fig. Helical wheel projection of a gramicidin b -helix with residues R pointing outward due to alternating D- and L-amino acid conformations in a b -sheet structure
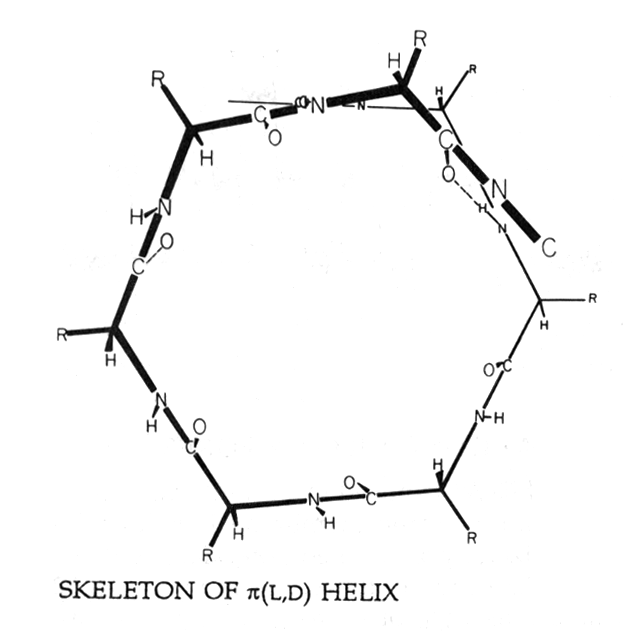
(from Hille, 1992)
Na+ ions moving through a Gramicidin pore carry along a single file of water molecules. Such a flux of ion and water molecules is called flux coupling because an ion and a water molecule can not pass each other within the channel. In the presence of a second type of permeable ion, the two ions couple their flux as well. A molecular dynamics simulation of ions moving through a Gramicidin dimer clearly shows the comparably fast movement of the peptide groups (calculation in vacuo). The ion moves back and forth in a zig-zag random walk driven by thermal motion. Considering that the channel allows for the flux of 107 ions per second, the molecular dynamics simulation nicely demonstrates the importance of movement of the peptide structure that occur at a time scale several order of magnitudes faster than the time required for the ion to cross the membrane. This means that the ion in contact with the peptide encounters a time averaged, static backbone structure.
This difference in time scale of substrate diffusion and protein motion is important for all enzymatic activities as shown for hemoglobin, where the static structures of the T and R state show no energetically favorable pathway for oxygen to diffuse to and away from the heme group. Only together with the rapid sidechain motions a pathway will be created which likely provides a mechanism to control substrate affinity.
Gramicidin channels have been studied electrophysiologically for over 30 years in planar lipid bilayers. The figure below is a single channel current recording of a synthetic membrane containing four active Gramicidin D unit channels. The recording (left) shows the baseline current and subsequent opening and closing steps of individual channels. Each upward transition reflects a dimerization event (opening of channel) and downward transitions reflect dimer dissociation (channel closing).
Fig. Single channel recording of four Gramicidin D channel units
(dimers)
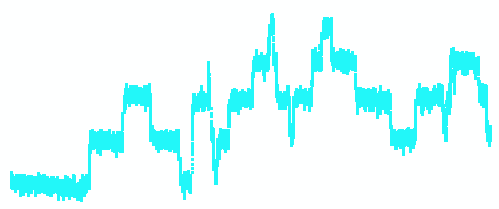
1. General
We commonly think that all we need to know about the building of an organism is its genetic blueprint - the genome. All the information is encoded in the DNA and the sequence of the genes coding for proteins therefore should contain all necessary information for the biosynthesis of a native protein. In other words, the sequence determines the 3-D structure. This idea spurred an experimental science to study the folding of proteins in vitro.
Proteins fold and unfold depending on the physical and chemical conditions of the environment. Protein folding can be studied in the test tube by dissolving a purified protein in aqueous solution and denature it using chaotropic agents which are polar and/or charged molecules. Removing this chaotropic molecules by dialysis induces a spontaneous refolding of the polypeptide into its original, native fold. The refolding experiment of proteins from the denatured, inactive to the native, active fold proves that the sequence determines the 3-D structure in aqueous solution. Protein folding is fast and occurs in multiple steps commonly referred to as folding pathway. To say that the sequence determines the native conformation is to say that the sequence determines the folding pathway that leads to a stable, functional 3-D structure.
Proteins are micelle-like structures that consist of a hydrophobic core and a hydrophilic surface (globular proteins). For membrane proteins the distribution of hydrophilic and hydrophobic amino acids is inverted in the membrane buried part. Most membrane proteins, however, have considerable extra-membranous domains with packing rules identical to globular proteins. Because membrane proteins need both hydrophilic and hydrophobic phases when solubilized in aqueous solution, membrane proteins are in general difficult to work with in isolated form and require the presence of detergents. Detergents can interfere with the folding properties of the membrane proteins and some are known to denature proteins. Working with membrane proteins therefore requires to find a suitable detergent that solubilizes the protein out of the membrane, yet keeps the native fold intact.
The studies of protein folding are entirely based on globular proteins. Like micelle formation, the folding of a globular protein is an entropically driven, spontaneous process but less so by the solvation energy of the contributing amino acid side chains. Folding of globular proteins is a good example of the hydrophobic effect. The denaturation of proteins by decreasing the temperature, cold denaturation, is caused by the weakening of the hydrophobic effect (the entropy term of solvation).
They change in free energy when going from the unfolded to the folded state D G(folded) ranges between -5 and -15 kcal/mol. This is called the stabilizing energy. This value has to be compared with the free energy of solvation of the folded or unfolded form of the protein which are in the order of -107 kcal/mol. Thus a relatively small stabilization energy is responsible for the stability of the folded over the unfolded state (D Gsol(folded) - D Gsol(unfolded) < 0).
2. Experimental procedure
Protein folding can be studied experimentally by following the folding pathway of the polypeptide chain. There are several spectroscopic methods available, including H1-NMR, fluorescence labeling, and circular dichroism (CD). Spectroscopic techniques exploit the changes in absorbance of individual amino acid residues when in contact with the solvent or adjacent amino acid residues. Secondary structural motif have an optical activity because they are regular patterns of optically active building blocks - the amino acids. Alpha helical and beta sheet content can be determined by circular dichroism. The CD spectrum changes with the amount of secondary structural elements and is therefore a suitable indicator of the folding state of a protein (although it doesn't distinguish native from non-native folds that have similar amounts and combinations of secondary structure).
A new, ultra-fast technique has been commented on in the journal Science
[Service, R.F., 1996]. Using laser technology, fluorescent tags indicate
the formation of helices in m s. The time resolution
in observing folding events has been a problem because of the lack of
synchrony among all protein units in the sample solution because they
do not start folding at the same time. Synchronization has now been achieved
by super-cooling the protein solution to -10°
C. A laser pulse of 8ns heats up the water by 20°
C and a second laser pulse of a few femto seconds (UV light) induces tryptophane
residues to emit a fluorescent signal. The intensity of the signal is
quenched by the proximity of methionine residues. Thus, by mutagenesis
of strategically located Trp and Met residues, the folding of secondary
structure elements during the first m s can
be followed experimentally.
3. Folding pathway
A general folding pathway can be described that includes 6 steps as shown
for the hypothetical folding of a dimeric protein.
| Pathway intermediate | Structural state |
| nucleation | transition of random coil to 2° elements secondary structure formation |
| condensation | growth and association between 2° elements into domains, cooperativity |
| molten globule | domains formed, but native 3° not achieved; disordered with hydrophobic residues partially exposed to water |
| 3° D | single polypeptide or subunit |
| 4° D | subunits assemble (dimer formation) |
| global energy minimum | native conformation |
4. Model systems
A few proteins served as model system for folding studies in vitro. These are the bovine pancreatic trypsin inhibitor (BPTI), Ribonuclease A, and T4 Lysozyme. They all have the following in common: their high solubility and great abundance (easy to purify), their structure has been solved, they have a testable enzymatic activity, and they contain disulfide bridges. For a refolding experiment they can be used to stabilize and study folding intermediates as summarized in the above table. These intermediate states are important for an understanding of those structural parts which in a protein fold appear first and therefore determine the overall folding process. Protein folding is a process of nucleation and 'aggregation' that shows cooperativity in the interaction of secondary structural elements. What is the experimental evidence? Here is a refolding experiment using Ribonuclease A. Increasing the temperature of the Ribonuclease A solution above 70° C changes the optical rotation of polarized light (as measured at 366nm). The denaturation curve shows a sharp transition indicating that the unfolding process is a cooperative process. The midpoint of the transition is called the melting temperature, Tm. The melting temperature for Ribonuclease A is 61.2° C. The denaturation is reversible and Ribonuclease A can successfully be renatured into an active fold upon slow cooling of the protein solution.
The great success of studying folding intermediates by following disulfide bridge formation came from the observation that the wrong conformations can often be found during folding. Intuitively a protein could search for the native fold by going through all possible conformations (in the most extreme scenario). Yet this is not the case. During folding, a protein often goes along an incorrect pathway, but it finds its way back to the correct one. Exactly how the sequence determines the 3° structure is not fully understood.
Experiments with T4 Lysozyme have shown which factors contribute most
to the stability of this protein:
- hydrophobic effect
- H-bonds
- Van der Waals interaction
- electrostatic
- helix dipole
- SS bridge
It is not intuitively obvious why disulfide bridges are not strong factors
in stabilizing protein folds. One of the conceptual difficulties is to
misunderstand that there exists a plethora of unfolded states that are
not necessarily elongated chain conformations, but can adopt any kind
of secondary structure nucleation. Also disulfides need not to be reduced
for a protein to be considered denatured. It has been shown experimentally
that SS bridges are somewhat limited in stabilizing a protein structure
because their formation is critically dependent on the distance (allowed
values 4-7.5Å) between the 2 a -carbon
centers of the cystein residues of adjacent peptide backbones. Disulfide
formation will not occur if backbone-to-backbone distance and orientation
of the cysteins are not optimal. Using site directed mutagenesis to introduce
new SS bridges often results in a destabilized protein (lower Tm)
because the rigid disulfide bond distorts the polypeptide backbone. In
addition, disulfide bonds have no role in early folding because even though
they covalently link neighboring parts of the polypeptide, they themselves
do not determine which parts.
In general, destabilization occurs at residues with low side chain mobility
and those which are inaccessible to solvent in the folded state. Unpaired
hydrogen bonds (and unpaired charged groups) destabilize the structure
because of the loss of the interaction with water in the unfolded state.
Thus individual water molecules within the protein can stabilize the fold,
if they can hydrogen bond with hydrogen bond donors or acceptors on the
protein.