Structure Determination Techniques
1. General
Protein secondary structure, DNA double helix and RNA stem and loop structures, the structures of lipids, membranes and polysaccharides are not simply derived from first principles, rather the principles have been extracted and confirmed from structure determination of the molecules at hand. There are many techniques to study different aspects of structures of cellular components, but two techniques allow a resolution at the level of distinguishing individual atoms: X-ray crystallography and Nuclear Magnetic Resonance or NMR technique.
X-ray crystallography has been used to determine the structure of inorganic and organic crystals since the early years of this century. The technique was first used for the elucidation of salt crystal structure, which for example gave Linus Pauling the instrumentation to study atomic distances from which he developed his theory of the chemical bond (combining structural information with quantum mechanical calculations). From the knowledge obtained from salt crystals Pauling, who focused his attention on protein structures, proposed the alpha helical and beta strand secondary structures [Pauling and Corey, 1951. Atomic coordinates and structure factors for two helical configurations of polypeptide chains], both of which have been confirmed by X-ray crystallographic analysis the first time using crystals of myoglobin and hemoglobin in the early 60ies by Kendrew and co-workers [Kendrew et al., 1960. Structure of myoglobin. A 3-dimensional Fourier synthesis at 2Å resolution. Nature 185:422-427].
X-ray structures are high resolution structures enabling the distinction of two points in space as close as 2Å apart. Yet they depict a static structure, the result of a technique which requires large, stable protein crystals, within which each protein unit is lined up in a regular lattice. It was soon recognized that these static structures didn't really help explaining function because the structures are mostly the average of millions of identical units. 'Loose' structural parts like surface loops often failed to be resolved leaving some protein structures incomplete. The development of nuclear magnetic resonance techniques, NMR, could be used to overcome this problem. In contrast to protein crystals needed for X-ray diffraction, NMR made use of protein solutions allowing for the determination of structures at very short time ranges. Consequently those flexible loop and domain structures could be solved successfully.
Today also cryo electron microscopy (a technique of using extremely low
temperature and rapidly freezing of samples) allows for a resolution of
well ordered sheets of protein complexes of as little as 5-10Å.
The resulting electronmicrographs show quaternary structure arrangements
of macromolecular complexes. Combining high resolution structures of protein
domains and subunits with electronmicrographs by superposition of the
two distinct structures yields an immense wealth of new structural information.
2. X-ray crystallography
X-ray crystallography makes use of the diffraction pattern of X-rays that are shot through an object. The pattern is determined by the electron density within the crystal. The diffraction is the result of an interaction with the high energy X-rays and the electrons in the atom. The electrons get activated and their relaxation to the initial energy state emits new X-rays. Bundles of such waves can be enhanced if they are in phase, and they get canceled out if they are out of phase. Therefore the diffraction of parallel X-rays from an object containing thousands of unit molecules arranged in a regular lattice results in the enhancement and cancellation of the diffracted waves and a resulting pattern of this vectorial process can be correlated with the distribution of the electrons in the crystal.
X-ray crystallography requires the growth of protein crystals up to 1 mm in size from a highly purified protein source. Crystal growth is an experimental technique and there exists no rules about the optimal conditions for a protein solution to result in a good protein crystal. The protocol has to be established for every new type of protein. Water soluble proteins are easier to crystallize than membrane proteins. The latter tend to precipitate out of solution due to unfavorable protein-protein and protein-solute interactions. To be kept soluble in aqueous solution, membrane proteins need the addition of detergents. The presence of detergents, however, often interferes with regular arrangements of the protein complexes in the crystal resulting in diffuse diffraction pattern. If membrane proteins contain large extra-membranous domains, these water soluble domains can be cleaved off from the membrane buried domain and crystallized individually.
X-rays have a wavelength of 0.2Å to 2.0Å. The wave length, as in an optical microscope, determines the resolution limit of half the applied wave length. X-rays are therefore suited for the atomic distances which reside in the angstrom range. X-rays are high energy electromagnetic radiation and can be recorded on X-ray sensitive film, the normal technique to record diffraction patterns of protein crystals.
X-rays that interact with an electron cause it to oscillate. Oscillating electrons serve as a new source of X-rays that propagate away from the stimulated electron. The waves of neighboring electrons super impose and depending on their being in-phase or out of phase result in a signal or in no signal at all. Diffraction by a crystal can be regarded as the reflection of the primary beam by sets of parallel planes that define the dimensions of the unit cell (the smallest repetitive pattern) of the crystal. The relationship between reflection angle, Q, the distance between the planes, d, and the wavelength, l, is given by Bragg's law:
2dsinQ = l Bragg's Law
The 2-dimensional distribution of the diffraction pattern can be calculated back into a 3-dimensional space of the electron distribution causing the diffraction. The mathematical formalism to do this is called Fourier transformation. The distances between the spots inversely correlates with the distances of the unit cell in the crystal and the intensity of the spots with the density of electrons in the molecular structure. The exact location of the electrons, however, is lost in a single diffraction pattern, because the information of the phase of the diffracted beams is not given. This is called the phase problem and is the hardest obstacle to overcome. The phase problem requires at least 3 different protein crystals with identical unit cell geometry and the inclusion of evenly spaced heavy metals or derivatives in the protein structure that give information about the relative phase in the individual crystal. The diffraction spots originating from the electron shell of the heavy metals can easily be identified and distinguished from other electron dens centers in the crystal. From the heavy metal location in the unit cell and the phase shift can be determined. The method to solve the phase problem using different crystals with identical protein structures containing regularly but infrequently spaced heavy metals or protein isoforms is known as multiple isomorphous replacement.
The amplitudes and phases of the diffraction data are used to calculate an electron-density map of the repeating unit of the crystal. This is a step that involves the interpretation of the raw data. This step is sensitive to the resolution of the diffraction data, which in turn is determined by the quality of the protein crystal, i.e., the regularity of the lattice of the protein in the unit cell and the regularity of the distribution of the heavy atom inclusions. The interpretation of the diffraction data needs information about the amino acid sequence of the protein because depending on the resolution of the data different amino acids can have indistinguishable electron densities (e.g. Tyr and Phe, or Leu and Ile).
Initial models of protein structures due to limits in the resolution
have to be refined. This is often achieved by comparing the experimental
data with the optimal structure obtained by computer modeling. The difference
in experimental structure and hypothetical structure is given as R-factor.
3. Nuclear magnetic resonance, or NMR
Nuclear magnet resonance obtains the same high resolution using a very
different strategy. NMR measures the distances between atomic nuclei,
rather than the electron density in a molecule. With NMR, a strong,
high frequency magnetic field stimulates atomic nuclei of the isotopes
H-1, D-2, C-13, or N-15 (they have a magnetic spin) and measures the frequency
of the magnetic field of the atomic nuclei during its oscillation period
back to the initial state. The important step is to determine which resonance
comes from which spin. The distance and type of neighboring nuclei determines
the resonance frequency of the stimulated atomic nuclei. This dependence
on next neighbors known as chemical shift (or spin-spin coupling
constant) andreflects the local electronic environment and the information
contained in 1-D NMR spectra. For proteins, NMR usually measures
the spin of protons. The following reasons make the H-1 NMR spectroscopy
the method of choice for biological macromolecules:
- H are present at many sites in proteins, nucleic acids, and polysaccharides- H have a high abundance for each site
- H nuclei is the most sensitive to detect
1-D spectra contain the information about all the chemical shifts of all
the H in the protein. The frequency resolution is often not enough to
distinguish individual chemical shifts. 2-D NMR solves this problems by
containing information about the relative position of H in molecular structures.
2-D NMR spectra contain information about interaction between H that are
covalently linked through one or two other atoms (COSY or correlation
spectroscopy). Alternatively, pairs of H that can be close in space,
even if they are from residues that are not close in sequence (NOE spectra,
or Nuclear Overhauser Effect). A complete structure can thus be
calculated by sequentially assigning cross peak correlations in 2-D spectras.
Currently, the size limit for proteins amenable to NMR solution structure
analysis is about 200 amino acids. An important feature of the identification
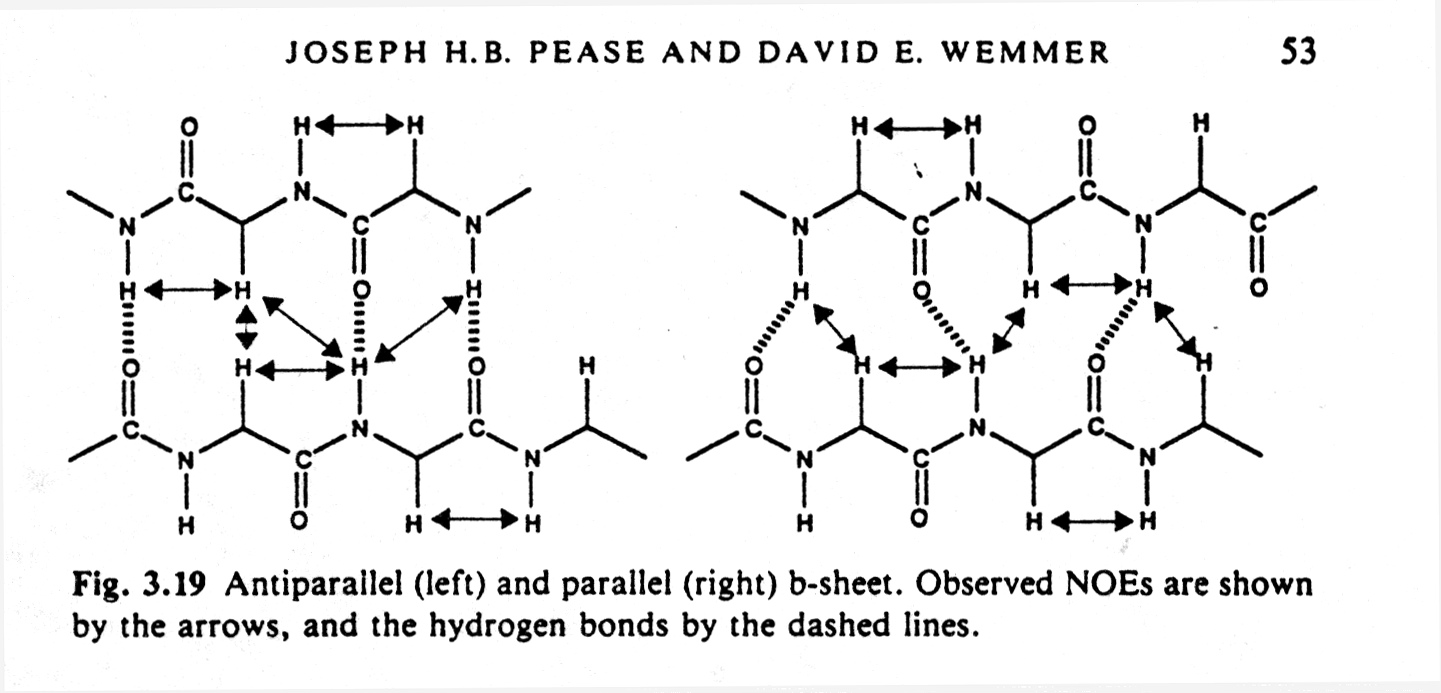
of cross peaks is that regular patterns can be recognized that stem from
secondary structure elements such as alpha helices and parallel or anti-parallel
beta sheets because they contain typical hydrogen bonding networks.
Fig. Observed NOEs in antiparallel and parallel b sheets
NMR also requires the knowledge of the amino acid sequence, but
the protein does not have to be in an ordered crystal, yet high concentrations
of solubilized protein must be available (NMR structures are therefor
also called solution structures). In biopolymers, the primary structure
(sequence) logically breaks up the molecule into groups of coupled spins
normally one or two groups per residue. This is true not only for proteins,
but also for nucleic acids and polysaccharides.
4. X-ray crystallography and NMR are complementary techniques
| NMR | X-ray crystallography |
| short time scale, protein folding | long time scale, static structure |
| solution, purity | single crystal, purity |
| < 20kD, domain | any size, domain, complex |
| functional active site | active or inactive |
| domains | domains |
| atomic nuclei, chemical bonds | electron density |
| resolution limit 2-3.5Å | resolution limit 2-3.5Å |
| primary structure must be known | primary structure must be know (except if resolution is 2Å or better for every single residue) |